
Piloting 168极速赛车开奖结果 全国统一一分钟极速赛车开奖结果 opportunities for our customers
from 168娱乐网·极速赛车一分钟开奖平台 lab tests to industrial-scale trials




Latest news
- Sep 26, 2024Record and payment dates for Valmet’s second dividend instalment for year 2023
- Sep 26, 2024Valmet to deliver a quality control system for corrugator to Inter Eastern Container in Thailand
- Sep 24, 2024Inside information: Valmet to supply a complete pulp mill with full-scope automation and flow control solutions to Arauco in Brazil
- Sep 24, 2024Change in Valmet's Executive Team
Explore our recent references around the world



Industries we serve 一分钟极速赛车官方开奖结果 娱乐168体彩官网开【极速赛车】开奖记录+历史结果

Pulp
Valmet’s fiber technical know-how provides intelligent, integrated and complete processes for both chemical and mechanical pulping. Our portfolio is built on strong R&D to guarantee high end-product quality. For improved process efficiency and increased business profitability, turn to Valmet. We offer intelligent automation solutions and services for the pulping industry, with solid experience and references.
Read more
Board and paper
Valmet’s expertise is rooted in experience. We’ve been co-operating with our customers in more than 700 board machine and 900 paper machine deliveries worldwide. Our offering includes everything for profitable board and paper production: innovative technologies, reliability and performance adding services as well as advanced automation solutions to guarantee that your paper machine runs smoothly, energy-efficiently and uses raw materials economically.
Read more
Tissue
Valmet's flexible tissue making technology provides sustainable production of all types of grades from plain to textured and structured tissue products with high quality. But we are not only limited to be a supplier of innovative solutions for the entire mill. We support you in planning, realization, installation and training as well as during the entire life cycle in order to make sure the mill operates at its optimum over the years.
Read more
Energy
Valmet is a reliable technology partner in the ever-changing energy markets. Based on our decades-long experience, we have the know-how to deliver energy solutions based on biomass, waste or on a mixture of different fuels. Together we can develop innovative, tailored solutions from our wide technology, automation, and services offering.
Read more
Valmet's climate program − Forward to a carbon neutral future
Valmet's climate program − Forward to a carbon neutral future − includes ambitious CO₂ emission reduction targets and concrete actions for the whole value chain, including the supply chain, our own operations, and customers’ use of our technologies.
Read more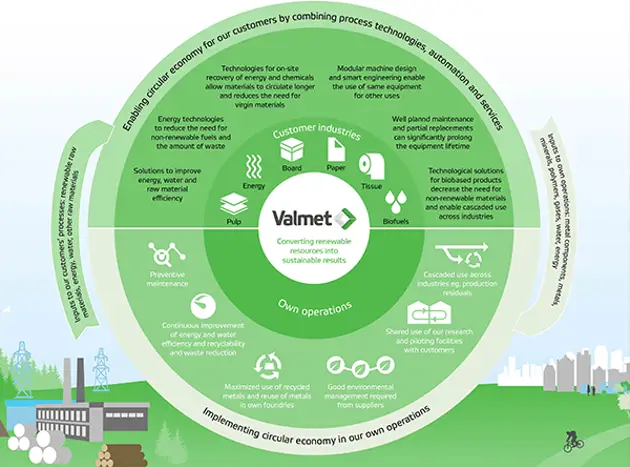
Taking the circular economy forward
Valmet is well positioned regarding circular economy. With our technology and services, we enable our customers to produce sustainable products from renewable materials and at the same time we continuously develop our own processes.
Read more
When 一分钟的极速开奖平台 168极速赛车开奖结果 官方网历史开奖记录查询168赛车七五秒直播 everything works together
Valmet is where the best talent from a wide variety of backroands comes together. Our commitment to moving our customers' performance forward requires creativity, technological innovations, project excellence, service know-how - and above all, teamwork.
Explore Careers at Valmet